Research Grants
Program
Novel research by leading experts funded through PSC Partners

Research Grants
Funding Milestones
2007
PSC Partners launched its Research Grants program
2009
Awarded $40,000 grants to Mayo Clinic, UC-Davis and the Adademic Medical Center in the Netherlands
2010
Over $500,000 had been awarded in grants.
2016
PSC Partners Canada joined with us to fully fund their first research grant
2018
Launched Young Investigator Award with PSC Partners Canada to fund up to $80,000 for promising researchers
2021
PSC Partners reached the $1 million funding milestone
2021
Award amounts increased to $100,000 for Young Investigators and $75,000 for Standard Seed Grants.
Leading the Way to a Cure
Since 2007, PSC Partners has awarded over $5 million for groundbreaking research.
117
1
$5.15 M
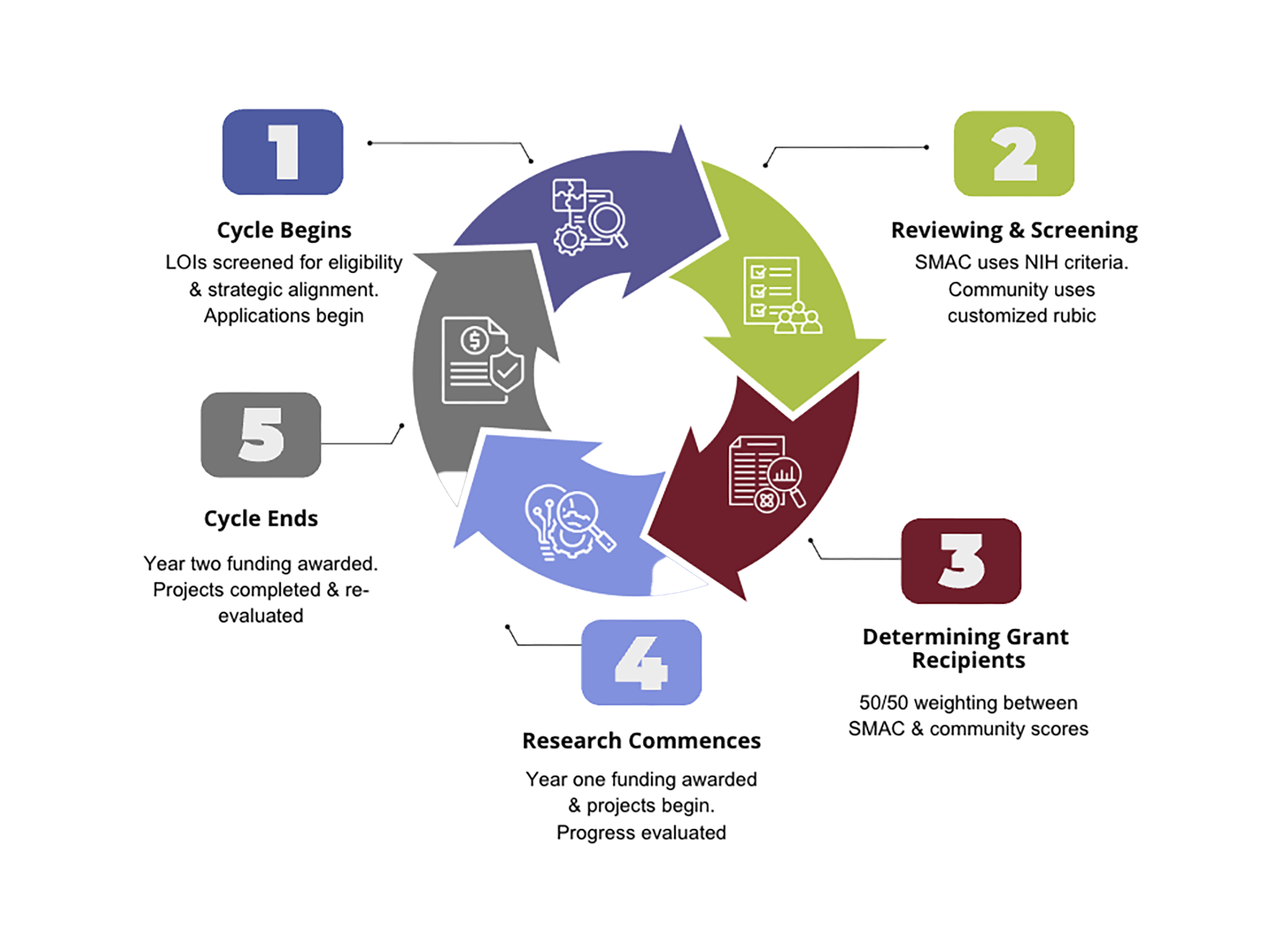
Grants Cycle
The grants program has an annual cycle, starting with letters of intent (LOIs) and progressing to applications, reviews, funding decisions, and reporting.
Grant Reporting Requirements
- PSC Partners staff conduct internal assessments on the grants program’s impact, progress, and areas for growth.
- Grant recipients provide a lay summary for the PSC Partners website and a first-year progress report for SMAC to review.
- Second-year funding is contingent upon a successful review of the progress report.
- Grant recipients complete the 2nd year of the research project and submit a final report.
PSC Partners Annual Research Grant Awards
Every year, based on recommendations from the Scientific Medical Advisory Committee (SMAC) and Community Reviewers, the Board of Directors of PSC Partners reviews all applications and funds the most promising PSC-related studies. In 2024, we announced funding for four international studies, and our affiliate PSC Partners Canada announced funding for one Canadian study. PSC Partners and affiliate PSC Partners Canada offer grants to conduct research that addresses an important and novel, basic or clinical research question related to PSC and closely associated diseases (such as inflammatory bowel diseases (IBD) and cholangiocarcinoma). Our Research Grants Program seeks to encourage investigators to conduct research in promising new areas, with the goal that data generated will lead to federal (NIH) or external international funding.
For more information, email grants@pscpartners.org.
